the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 19 Dec 2019
| 19 Dec 2019
Identifying and quantifying geogenic organic carbon in soils – the case of graphite
Jeroen H. T. Zethof
Martin Leue
Cordula Vogel
Shane W. Stoner
Karsten Kalbitz
A widely overlooked source of carbon (C) in the soil environment is organic carbon (OC) of geogenic origin, e.g. graphite, occurring mostly in metamorphic rocks. Appropriate methods are not available to quantify graphite and to differentiate it from other organic and inorganic C sources in soils. This methodological shortcoming also complicates studies on OC in soils formed on graphite-containing bedrock because of the unknown contribution of a very different soil OC source.
In this study, we examined Fourier-transform infrared (FTIR) spectroscopy, thermogravimetric analysis (TGA) and the smart combustion method for their ability to identify and quantify graphitic C in soils. For this purpose, several artificial soil samples with graphite, CaCO3 and plant litter as the usual C components were created. A graphitic standard was mixed with pure quartz and a natural soil for calibration and validation of the methods over a graphitic C range of 0.1 % to 4 %. Furthermore, rock and soil material from a graphite-bearing schist and a schist without natural graphite were used for method validation.
FTIR. As specific signal intensities of distinct graphite absorption bands were missing, calibration could only be performed on general effects of graphite contents on the energy transmitted through the samples. The use of samples from different mineral origins yielded significant matrix effects and hampered the prediction of geogenic graphite contents in soils.
TGA. Thermogravimetric analysis, based on changes in mass loss due to differences in thermal stabilities, is suggested as a useful method for graphite identification, although (calcium) carbonate and graphitic C have a similar thermal stability. However, the quantitative estimation of the graphite contents was challenging as dehydroxylation (mass loss) of a wide range of soil minerals occurs in a similar temperature range.
Smart combustion. The method is based on measuring the release of C during a combustion program, quantified by a non-dispersive infrared detector (NDIR) as part of a commercial elemental analyser, whereby carbonates and graphitic C could be separated by switching between oxic and anoxic conditions during thermal decomposition. Samples were heated to 400 ∘C under oxygen-rich conditions, after which further heating was done under anoxic conditions till 900 ∘C. The residual oxidizable carbon (ROC), hypothesized to be graphitic C, was measured by switching back to oxygenic conditions at 900 ∘C. Test samples showed promising results for quantifying graphitic C in soils. For the purpose of quantifying graphitic C content in soil samples, smart combustion was the most promising method of those which have been examined in this study. However, caution should be taken with carbonate-rich soils as increasing amounts of carbonate resulted in an underestimation of graphitic C content.
- Article
(2902 KB) - Full-text XML
-
Supplement
(774 KB) - BibTeX
- EndNote





In the past decades, global carbon (C) cycling has gained more and more attention. As an important component in this cycle, the soil C reservoir consists of many different forms and types of carbonaceous substances, each with unique turnover times and functions. Probably the most widely studied and dynamic soil C component is fresh organic matter (OM), derived from litter input, decaying organisms and plant exudates (summarized in Blankinship et al., 2018). Another well-studied soil C component is inorganic C in the form of carbonates, which forms an important part of the soil C pool, especially under arid climate conditions (Zamanian et al., 2016; Apesteguia et al., 2018). Black C, defined as a broad set of highly condensed carbonaceous by-products (e.g. soot) and residues (e.g. charcoal) of incomplete fossil fuel and biomass combustion, has obtained increasing interest during the past decades (Agarwal and Bucheli, 2011). Organic C (OC) of geogenic origin, which has gained less attention until now, is formed when organic compounds in sediments undergo coalification or kerogen transformation during diagenesis. Under high pressure and appropriate temperature conditions this process can continue into the formation of graphitic C, although well-crystallized pure C is rarely produced (Oohashi et al., 2012; Buseck and Beysacc, 2014). Redox transformations during metamorphoses of carbonates leads also to the formation of highly crystalline graphite (Galvez et al., 2013). Intruding hydrothermal fluids in the earth's crust forms a second source of graphitic C during rock formation, which produces the purest graphite crystals (Rumble, 2014). This relatively pure and stable form of C is highly chemical inert, although impurities from the parent material increase its chemical reactivity (Beyssac and Rumble, 2014). Via tectonic processes graphite bearing rocks can reach the earth's surface where they are subjected to physical and chemical weathering. Therefore, graphitic C occurs mainly in rocks from orogenic belts and in metasedimentary rocks in old cratons and might be a quite common bedrock for soil development (Hartmann and Moosdorf, 2012; Buseck and Beysacc, 2014).
The fate of geogenic graphite under weathering and soil formation has rarely been studied, possibly due to the lack of methods for determining and quantifying geogenic graphite beyond the background of soil OC. There are some indications that a substantial part of the geogenic graphitic C is actually lost in the pathway from rock weathering to (marine) sedimentation (Galy et al., 2008; Clark et al., 2017). Isolated naphthalene-degrading bacteria from contaminated soil proved to oxidize and degrade graphitic materials, questioning the assumed biological inactivity of graphite (Liu et al., 2015). In a recent study, Hemmingway et al. (2018) estimated that 2∕3 of the graphitic C is oxidized during soil formation, strongly facilitated by soil microbial activity.
The necessity of identifying and quantifying geogenic C becomes obvious when considering the widely used 14C dating method to measure the mean age of substances and their turnover rates (Trumbore, 2000). As C is depleted in 14C over 50 000 years following burial, geogenic C will contain no longer 14C and might dilute the 14C content of younger C pools (Rumpel and Kögler-Knabner, 2011). Although the dilution effect might be of less importance for the C pool in topsoil, it can become more important in subsoil as the C gets older and geogenic C might have a more dominant share in the total C pool (Rumpel and Kögler-Knabner, 2011). If geogenic C cannot be distinguished from the “normal” soil organic C derived from fresh OM, age and turnover time of soil OC will be overestimated. For instance, in the case of Hemmingway et al. (2018), the OM in the upper soil layers should have aged for over 20 000 years to explain the derived 14C signal, which is unlikely under the local tropical conditions. Although exact figures are unknown, more than one fifth of the global lithology may potentially contain graphitic C (Hartmann and Moosdorf, 2012). This illustrates the necessity to distinguish between the different C sources in order to study their fate and residence time in soils.
Several quantification methods, based on optical, thermal and chemical properties, have been established for identification and quantification of various C sources. Probably the most widely applied method is measuring C released after dry combustion. However, dry combustion is not suitable for differentiating between different (in)organic C components, which therefore have to be corrected for or even removed. Several pre-treatments, like thermal differentiation (e.g. Apesteguia et al., 2018) or removal by acid fumigation (e.g. Harris et al., 2001) in the case of carbonates, have been established.
Spectroscopic techniques, such as Fourier-transform infrared (FTIR) spectroscopy in the mid-infrared range (wavelengths 2.5–25 µm), are widely used to qualify organic and mineral matter in soils in terms of its functional groups (Smith, 1995; Parikh et al., 2014). Transmission FTIR yields highly resolved spectra with clearly separated absorption peaks, but requires sample dilution. In contrast, diffuse reflectance infrared Fourier transform (DRIFT) can be applied to undiluted soil samples (Reeves, 2003), in particular to determine OC contents of soils (e.g. Reeves et al., 2002; McCarty et al., 2002) via partial least squares regression (PLSR) (e.g. Janik et al., 1998). For employing PLSR, DRIFT spectra are calibrated by OC contents obtained with standard techniques such as dry combustion (e.g. Vohland et al., 2014).
Mid-infrared spectra from graphite show few absorption bands. Among the bands at wave numbers 2200, 1587, 1362, and 830 cm−1 (Friedel and Carson, 1971), the bands at 1587 and 868 cm−1 were attributed to optical lattice vibrational modes of graphite (Chung, 2002), while the other two bands cannot be assigned to specific functional groups. Tan et al. (2013) reported no prominent peak in FTIR spectra from pure graphite powder. However, in the case of oxidized or impure graphite, a number of infrared absorption bands assigned to C–OH (3400 cm−1), C=O (1729 cm−1), phenolic C–OH (1220 cm−1), C–O (1052 cm−1) and aromatic C–H (870 cm−1) have been reported (Tan et al., 2013). Depending on the graphite C amount and transformation stage, it has not been clear until now whether they can be defined in soil samples.
Thermal/thermogravimetric analyses (TGA) have been applied for a long time to study the mineral components of soils and rocks. For instance, the Rock-Eval method has been developed for oil and gas exploration, measuring the hydrocarbon, CO and CO2 concentrations by a consecutive pyrolysis and oxidation program under constant heating (Behar et al., 2001). More recent thermal analyses have been adopted to study the oxidative behaviour of soil OC, which might serve as a proxy for biogeochemical stability of these substances (Plante et al., 2009). The Rock-Eval method has been successfully applied to characterize the more stable part of OM remaining in the soil after long-term bare fallow (Barré et al., 2016). One of the advantages of TGA is the relatively inexpensive approach with minimal sample preparation needed to distinguish between different soil C components (Plant et al., 2009; Fernández et al., 2012; Kučerík et al., 2018). Additionally, it is a promising method to differentiate between the thermally instable OM and highly stable geogenic C, like coal or graphite. A precondition for quantifying substances with the TGA method is that the thermal properties of the substance of interest are known, i.e. the temperature limits at which the oxidation ∕ decomposition reactions take place. As no universally accepted temperature limits currently exists, the method still depends on empirically derived temperature boundaries to differentiate and quantify substances (Pallasser et al., 2013; Ussiri et al., 2014).
Fernández et al. (2012) demonstrated that TGA coupled with differential scanning calorimetry and evolved gas analysis (CO2∕H2O) increases the accuracy of quantifying organic substances during thermal analysis. In particular, the detection limit of CaCO3 could be decreased compared to conventional TGA since the decomposition could be masked by the dihydroxylation of (clay) minerals (Fernández et al., 2012). The same principle can also be expected for oxidation of graphitic C, as it takes place at roughly the same temperature ranges as the (clay) dihydroxylation and CaCO3 decomposition (Hayhurst and Parmar, 1998; Bews et al., 2001).
Recently, a new method has been developed based on experience with TGA measurements, which is defined in the DIN19539 standard (DIN Standards Committee Water Practice, 2016). In short, the DIN standard defines biologically labile OM in solid samples, including soils, to be thermally oxidized at temperatures below 400 ∘C (TOC400), while residual oxidizable C (ROC), like lignite or soot, and inorganic C (TIC900) are oxidized or decomposed between 400 and 900 ∘C. Combustion elemental analysers, based on this DIN standard, also offer the possibility of alternating between oxic and anoxic conditions during a measurement. In this method, named “smart combustion”, C components are consequently differentiated on both thermal and oxidizable properties. In theory, graphite as a pure C will oxidize poorly under anoxic conditions (Hayhurst and Parmar, 1998; Bews et al., 2001), while carbonates do not require oxygen to decompose at these temperatures. In contrast to the pyrolysing step of OM by the Rock-Eval method (Behar et al., 2001), OM is immediately oxidized in the first heating phase with the smart combustion method. Therefore, it is less likely that by-products of OM pyrolysis will end up in the same fraction as graphitic C.
In summary, graphitic C content in soils has received very little attention as a quantification method is lacking. This study aims to test several available methods for identifying and quantifying graphitic C content of soils by examining Fourier Transform Infrared spectroscopy (FTIR), thermogravimetric analysis (TGA) and the smart combustion methods. To test the validity of the above methods for graphite identification and quantification, we analysed natural and artificial soils that included widely present soil C components, i.e. carbonates and OM.
2.1 Artificial mixtures and soil/rock samples
Top soil and fresh rock samples from a nearby outcrop were taken from a field site in Rambla Honda, Sierra de los Filabres (37∘7′43′′ N, 2∘22′30′′ W/Southern Spain). The area is located in the Nevado-Filabride complex and contains Devonian-Carboniferous slaty mica schist with graphite and garnets crossed by abundant quartz veins (Puigdefábregas et al., 1996). Carbonates found in the soil sample (0.18 % C) originated from pedogenesis and dust deposition, as the parent rock does not contain carbonates. Soil material was taken from the topsoil (0–5 cm, without sieving crust) under the grass tussock Macrochloa tenacissima to ensure a substantial amount of OC was present. The soil itself was classified as Skeletic Leptosol (colluvic) according to the World Reference Base for Soil Resources (WRB, 2014). Additional soil material was collected from a field near the town of Alboloduy (37∘4′9′′ N, 2∘36′43′′ W), hereafter referred to as AB soil, with similar vegetation and (climatic) conditions. The lithology consists of feldspathic mica schist (IGME, 1979), but without natural graphite and with a much higher CaCO3 content (1.87 % C). The AB soil, classified as Skeletic Leptosol (WRB, 2014), was also sampled from the topsoil, without sieving crust, under the grass tussock Macrochloa tenacissima. The soil samples were dried at 40 ∘C and sieved to ≤2 mm.
Furthermore, three artificial soils were created, resembling a simplified version of a natural soil sample. For artificial soil 1 the organic horizon under deciduous tree species in Tharandter Wald (Saxony, Germany) was collected as OM substitute. Muscovite (American Educational, PN B00657LD62), a primary mineral present in the collected rock and soil samples (IGME, 1975), was taken as mineral component and ground in an agate disc mill. Together with CaCO3 (Merck, Darmstadt, PN 1.02066) and graphite standard material (Merck, Darmstadt PN 1.04206), the components were mixed in a pure quartz matrix (Merck, Darmstadt, PN 1.07536) in the ratio of 10 % Muscovite, 2.4 % OM (=1.0 % C), 2.1 % CaCO3 (= 0.25 % C) and 0.5 % graphite (=0.5 % C). Additionally, the individual components were mixed in quartz in the same concentration. Artificial soil 2 was made without carbonate and Artificial soil 3 without graphite, whereby concentrations of the other components were kept constant (Table 1).
Table 1Overview of the used samples. Total carbon (TC) and total inorganic carbon (TIC) data are derived by the elemental analyser, as described in Sect. 2.1. Crosses note for which method testing the sample has been used.
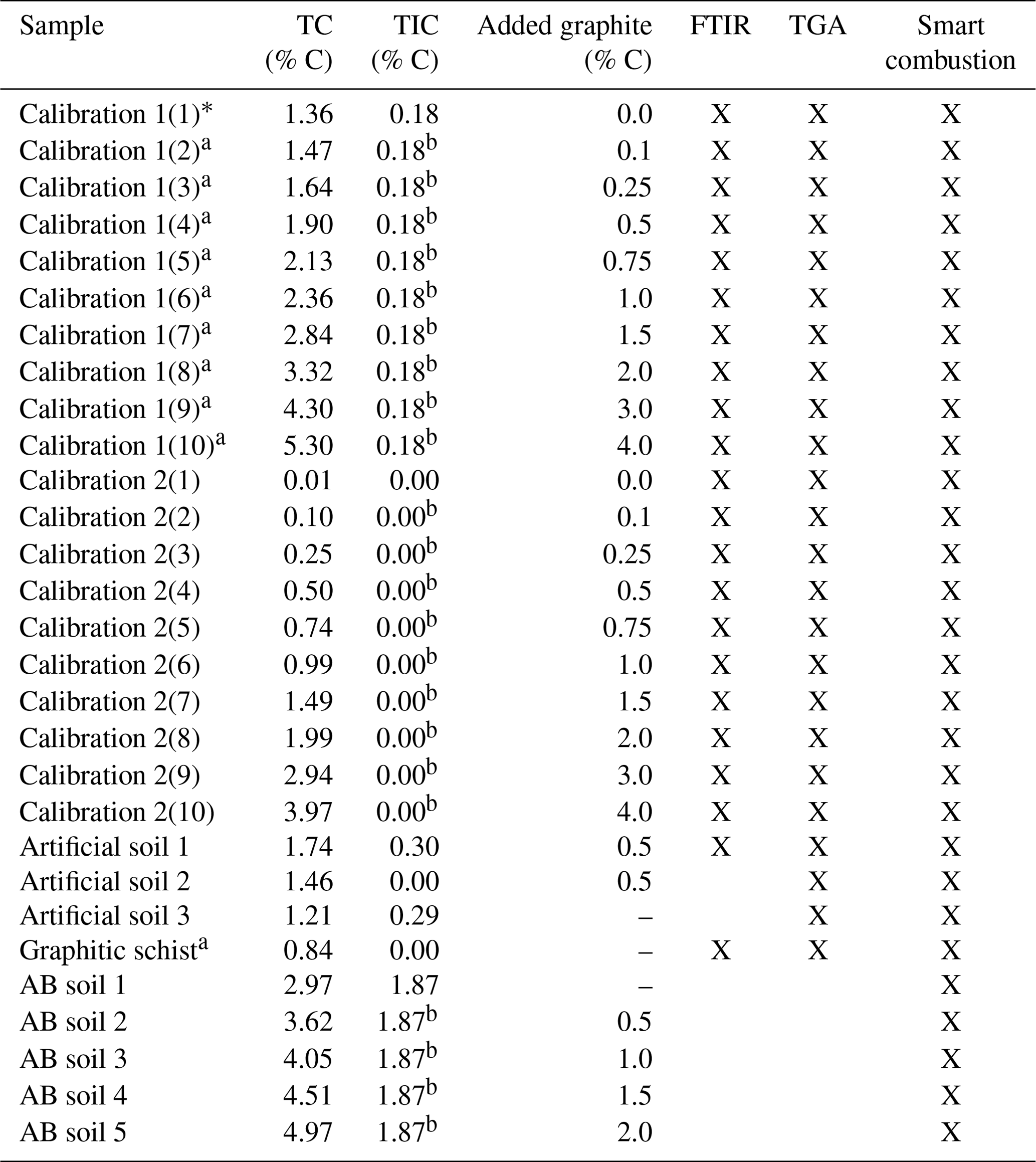
a Contain unknown amount of natural graphite. b As measured in the sample with 0.0 added graphite.
For testing and developing potential quantitative methods, the graphite standard was used as the reference material and added to soil from Rambla Honda (denoted calibration set 1) or with pure quartz (denoted calibration set 2) as a matrix in different quantities from 0.1 % to 4 % (Table 1). All samples were ground in an agate disc mill (Retsch GmbH, Haan, Germany) in order to achieve homogenization.
Total C content of the different mixtures and samples were measured and checked using an elemental CN analyser (Vario EL Cube, Elementar, Langenselbold, Germany). By acidifying samples, using an excess of HCl, the carbonates were removed. After drying at 60 ∘C, the difference between acid and non-acid treatment was measured and denoted as total inorganic C (TIC). TC and TIC values of the used samples are summarized in Table 1. We assumed that with dry combustion at 950 ∘C under pure oxygen atmosphere, all C holding components were decomposed or oxidized and therefore the total C content could be measured. This assumption was later validated by performing TGA temperatures up to 1100 ∘C.
Raman spectra of the soil of calibration set 1, standard (Merck) and graphitic schist were recorded, using a Thermo Scientific DXR Smart Raman Spectrometer, with a 532 nm laser and a power output of 9 mW. Before the measurement, samples were pressed in aluminium cups. The obtained peaks were integrated using Lorentzian profiles fitting in Origin 2019.
The spectra of the graphite standard were highly similar to the graphitic schist (Fig. S1 in the Supplement). The D1 (∼1350 cm−1), G (∼1580 cm−1) and D′ (∼1620 cm−1) peaks could be attributed to graphitic C. The ratio between the D1 and the sum of all three peaks are clear indicators for the degree of graphitization (Beysacc et al., 2003; Ferrari, 2007). Ratios of 0.20 for the graphite standard (Merck) and 0.34 for the graphitic schist indicated well-organized carbon (< 0.5, Beysacc et al., 2003) for both samples. Peaks around the 1200 and 1500 cm−1 bands, which would have indicated the presence of pyrogenic ∕ black carbon components (Sadezky et al., 2005; Schmidt et al., 2002), could not be observed in all samples, including the soil sample (blue, Fig. S1).

Figure 1Prediction plots with 95 % prediction bands (a, b) and loading plots (c, d) after FTIR analyses of the PLSR calibration sets using soil (calibration set 1) and pure quartz (calibration set 2) with graphite concentrations of 0.1 %–4 % added as indicated in Table 1.
2.2 Fourier transform infrared (FTIR) spectroscopy
For transmission-FTIR analyses, 1 mg sample was mixed with 99 mg potassium bromide (KBr; Merck, Darmstadt; 3 sample replicates), finely ground in an agate mortar, and pressed to pellets. The transmission spectra were recorded in a Biorad FTS 135 spectrometer (BIO-RAD company, Cambridge, USA) as 16 co-added scans between wave number (WN) 4000 and 400 cm−1 at a spectral resolution of 1 cm−1. The spectra were corrected against ambient air as background and were converted to absorption units. For DRIFT analyses, ground mixtures of calibration set 1 and 2 were poured into standard cups (three replicates) without any dilution. The DRIFT spectra (16 co-added scans, WN 4000 and 400 cm−1, resolution 4 cm−1) were corrected for ambient air using a background spectrum of a gold target (99 %; Infragold) and were converted to Kubelka–Munk units. All spectra were corrected for CO2 absorption of the ambient air between WN 2400 and 2280 cm−1 and smoothed (boxcar moving average algorithm, factor for transmission spectra: 25, factor for DRIFT spectra: 15) using the software WIN-IR Pro 3.4 (Digilab, MA, USA). For each sample one mean spectrum was calculated from the spectra of three replicate spectra.
2.2.1 Statistical analysis of the FTIR spectroscopic data
The partial least squares regression (PLSR) analyses of correlations between the transmission or DRIFT spectra and the graphite contents (0.1 %–4 %) of the samples were performed using R, Version 3.1.1 (R Core Team, 2014) with module PLS (SIMPLS, cross-validation: leave-one-out) of Mevik et al. (2018). The signal intensities were used as independent variables, the graphite content as dependent. The number of components used in the calibration models followed the lowest predicted root-mean-square error (RMSEP) of the specific data sets. The scores and loadings were plotted for the two main components determining most of the variances of the DRIFT spectra. Larger absolute loading values of signal intensities in certain WN regions imply a greater importance of these WN for the cumulated values of the principal components 1 or 2 displayed in the score plot.
2.3 Thermogravimetric analysis (TGA)
The TGAs were conducted on a STA 449 F5 Jupiter analyser (NETZSCH, Hanau, Germany). Therefore, 20–40 mg of sample material was placed in an Al2O3 crucible and heated under a constant heating program from ambient to 1100 ∘C with a ramp of 20 ∘C min−1. First analyses were conducted under an oxygen-rich atmosphere, with an inflow of 250 mL O2 min−1 and 250 mL N2 min−1. Additional tests were done under anoxic conditions whereby the oxygen inflow was cut off between 500 and 850 ∘C. The oxygen inflow was restored and the heating program continued until 1100 ∘C.
As carbonates might interfere in the TGA measurement of graphite and high chloride concentrations damages the equipment, they were removed from the sample using the acid fumigation method of Harris et al. (2001). Briefly, about 40 mg of sample was weighed in a silver foil capsules, moistened to approximately field capacity and put in a desiccator under vacuum conditions with a beaker of 31 % HCl and fumigated for 24 h. Afterwards the sample was dried at 60 ∘C overnight before it was transferred to an Al2O3 crucible for analysis.
2.3.1 Statistical analysis of the TGA method
TGA measurements were processed and thermal mass loss data obtained via the Proteus Thermal Analysis software (NETZSCH, Hanau, Germany). Initial testing with triplicates revealed that the repeatability error, expressed as coefficient of variation of the mass loss in areas of interest, was below 2 %. Measured sample mass data are fitted with the spline function over the temperature, with steps of 1 ∘C. Further analyses of the obtained data were conducted using R, Version 3.5.1 (R Core Team, 2018). Using the Fitting Linear Models function, models were created from the calibrations sets. With the module PLS (Mevik et al., 2018), a model was created for each calibration set to determine the graphite content based on the measured mass change in a certain temperature range and the known graphite addition, using the mass change as independent variable. By iterating the model creation over the temperature range from 400 to 1075 ∘C with minimum step of 5 ∘C difference and recording the slope, intercept and RMSEP of each created model, we evaluated the temperature range describing the graphite content of both the calibration sets most precisely. For this purpose, the RMSEP of these models were used (Fig. S4), and a single temperature range that fit both calibration sets was determined.
2.4 Smart combustion
Smart combustion denotes the method based on the DIN19539 (GS) standard (DIN Standards Committee Water Practice, 2016), whereby solid C components are separated based on their thermal and oxidizable properties. Smart combustion was conducted with the Soli-TOC cube analyser (Elementar, Langenselbold, Germany). The device is equipped with a non-dispersive infrared detector (NDIR), which measures the degree of infrared light absorbance caused by CO2 concentration in the measuring gas (O2∕N2). The NDIR was calibrated with CaCO3 and additionally CaCO3 was used to control and calculate a daily standard for the measured total C content. Depending on the expected carbonate and graphite concentration, 40–90 mg of homogenized sample was placed in the crucible. This was done to make sure that the peak surface fits well to the calibration range without causing unnecessarily large peak areas, which might influence the separation of the peaks/substances during analysis. Following the DIN19539 GS standard/standard gas switching program of the Soli-TOC cube analyser, the sample was first heated to and held at 400 ∘C for 240 s, whereby the “total organic carbon 400 ∘C” (TOC400) was obtained. Subsequently the atmosphere was switched to inert gas (N2) and after an equilibration time of 100 s the sample was heated to 900 ∘C and held for 150 s. C released during this pyrolysing phase is denoted as TIC, mainly consisting of carbonates which do not need oxygen to decompose. After 150 s, the oxygen gas flow was reintroduced, and a third C component, the residual oxidizable C (ROC), was measured. It was hypothesized that this ROC fraction represents graphite.
2.4.1 Statistical analysis of the smart combustion data
The Soli-TOC device directly converts the NDIR signal to C content of the different components, as calibrated with CaCO3. Creating an additional model to correct the C output introduces an additional error in the measurements. Therefore, we analysed the direct C output, as measured in the ROC fraction. Triplicate measurements were averaged, whereby the average coefficient of variation between replicates was 2.7 %. A Pearson correlation test was performed between the obtained ROC data and calibration sets to evaluate the graphite content measurements.
3.1 Overestimation of graphite contents by FTIR spectroscopy
The PLSR of the calibration set showed strong relations between the transmission-FTIR spectra from both calibration sets and the graphite concentrations when considering samples with 0.1 %–4 % graphite (Fig. 1a and b). For DRIFT spectra, the quality of these calibrations was at the same level (cal. Set 1: R2=0.97, RMSEP = 0.16; cal. Set 2: R2=0.98, RMSEP = 0.12). For calibration set 1 (based on natural soil) as well as for calibration set 2 (based on quartz), one main component of the PLSR presented most differences in the graphite concentration (Fig. 1c and d). This component showed the highest loading values across the entire range of wave numbers with some exceptions. For calibration set 1, wave numbers with decreased loading values were found at spectral regions 1077, 1031, 1013, 934, 913, 778, 536, 471, and 411 cm−1, which all corresponded to the prominent absorption bands of the original soil used as matrix, comprising functional groups from organic and mineral matter (Hesse et al., 1984; Senesi et al., 2003; Van der Marel and Beutelspacher, 1976). For calibration set 2, the wave numbers with the smallest loadings at 1171, 1084, 796, 778, 694, 506, and 457 cm−1 were specific for quartz (Van der Marel and Beutelspacher, 1976).
The cross-validation of PLSR calibrations 1 and 2 using spectra from the other calibration set yielded a linear relation but overestimated the graphite contents by ca. 2.59 % C for set 1 % and 1.87 % C for set 2 (Fig. 5). The graphite content of 0.50 % for the artificial soil was 3.5 times overestimated by the PLSR calibration using set 1 (predicted: 1.75 % C) and 3 times underestimated by calibration set 2 (predicted: 0.17 % C). The graphite contents in the graphitic schist were estimated to be 1.91 % C by calibration set 1 % C and 3.71 % C by calibration set 2, which was 2 times and 4.5 times higher than the total C content of the graphitic schist (Table 1).
3.2 Strong matrix dependency of TGA predictability
First qualitative TGA results revealed overlapping mass loss peaks of graphite and CaCO3 (Fig. 2). Between 750 and 850 ∘C, the sum of the mass losses of the individual components was smaller than the mass loss of the mixture of these components.
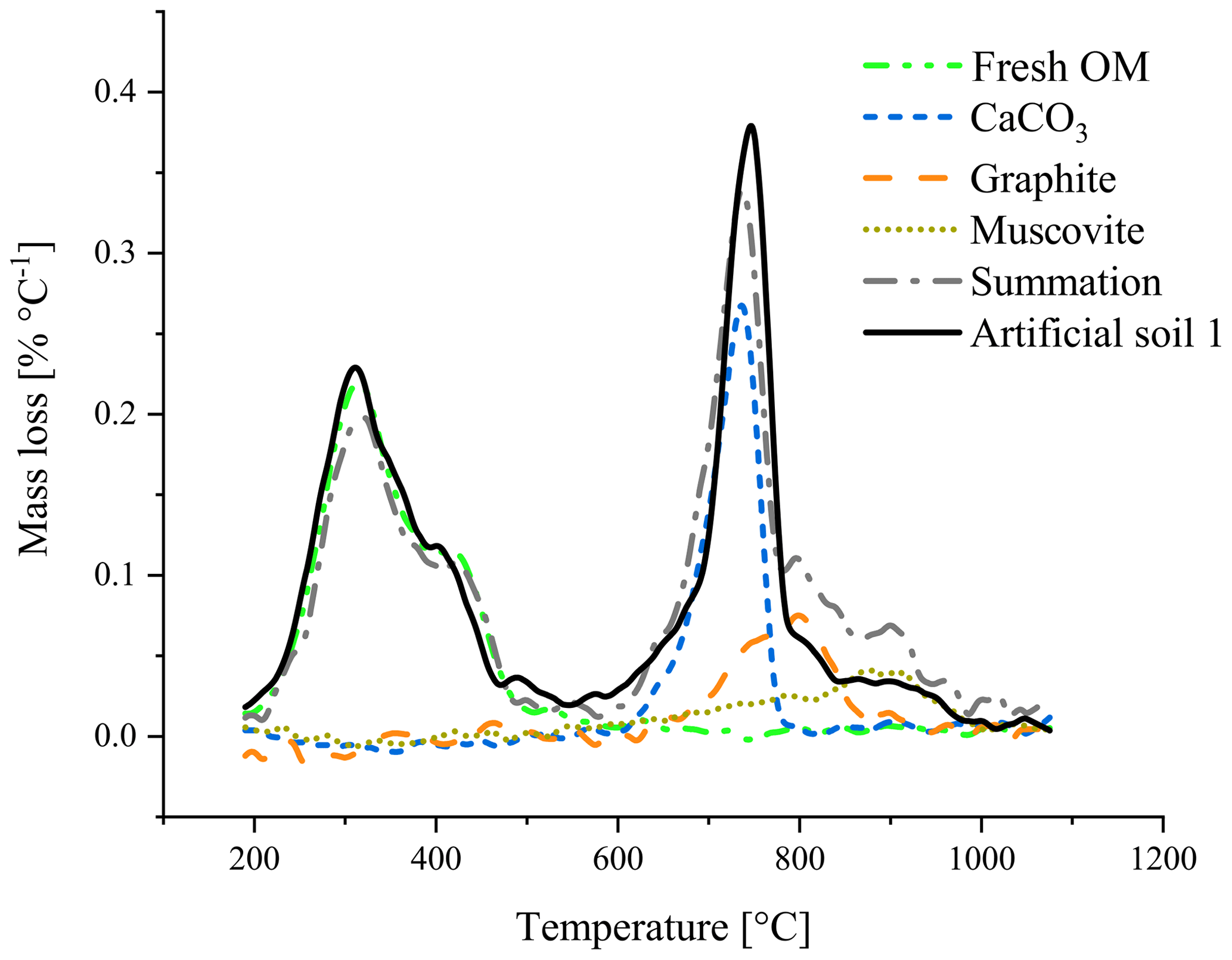
Figure 2Thermogravimetric analysis of artificial soil 1 and its components measured individually. The summation (dash-dotted, grey) is the combined mass loss of the individual components.
Using the RMSEP values of the two calibration sets of soil and quartz with graphite, the most useful temperature range for modelling graphite content was identified between 680 and 840 ∘C (Fig. S4). According to PLSR, for both models, created with calibration set 1 (R2=1.00, RMSEP = 0.05) and calibration set 2 (R2=1.00, RMSEP = 0.04), predicted graphite content in their respective matrix quite well (Fig. 3). Only cross-validation by predicting graphite content in the other matrix revealed a short coming of the TGA method (Fig. 5). The slope between predicted and actual graphite content is still parallel to the 1:1 line, but applying the model derived from calibration set 1 on set 2 underestimated the graphite content by 1.81 % C (Fig. 5). In contrast, the graphite content in calibration set 1 was underestimated by 1.81 % C using the model calibrated with set 2.
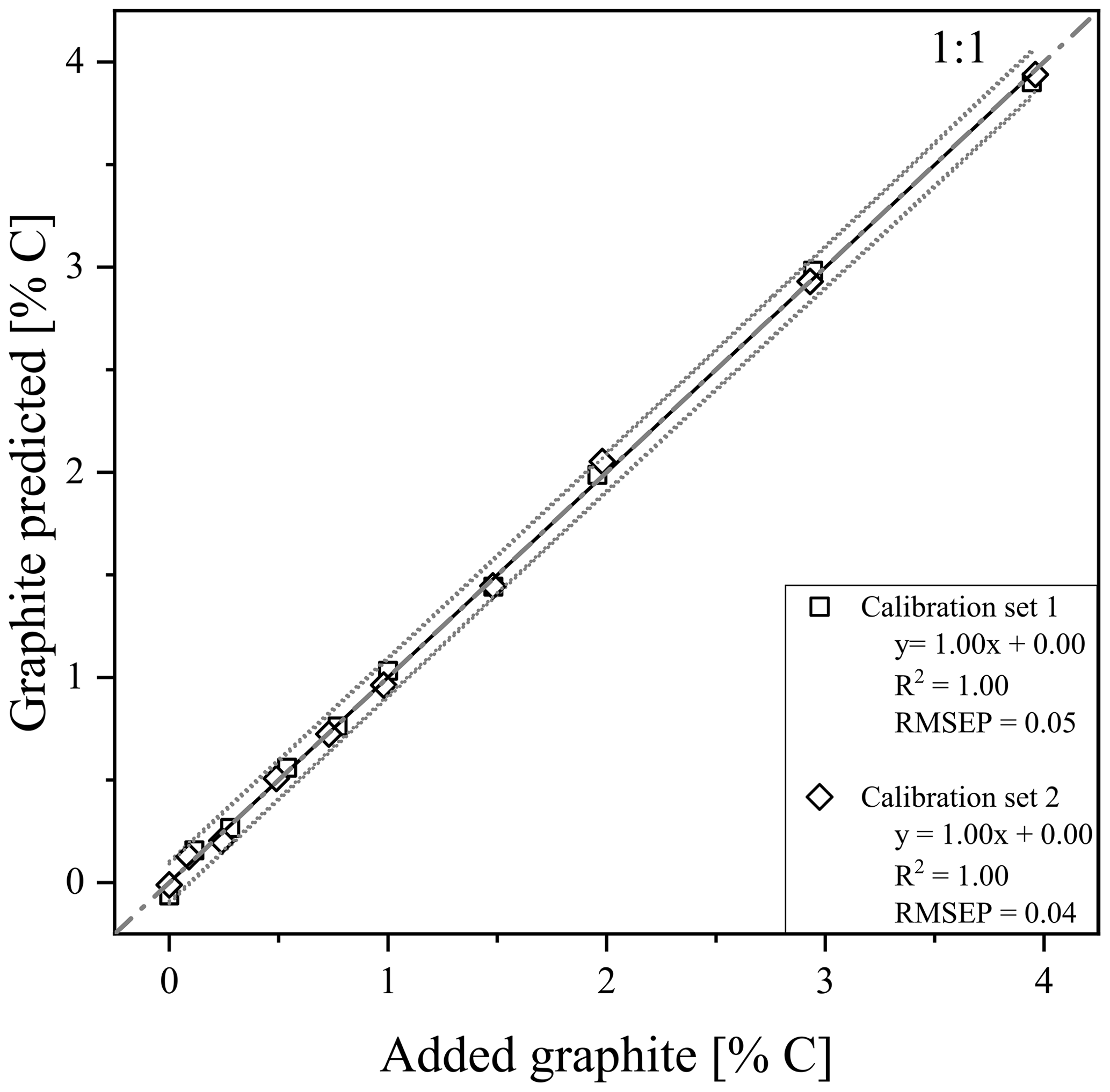
Figure 3Prediction plots after thermogravimetric analysis (TGA) of calibration set 1 (squares) and 2 (diamonds). 95 % predictions band (dotted grey) are displayed besides the linear regression line (black). Graphite was added in the concentrations between 0 % and 4.0 % graphite as indicated in Table 1.
The graphite contents of the artificial soil (0.5 %) and graphitic schist were estimated using the two calibrated models (Fig. 5, inserted detail, and Table 2). The content of artificial soil 1 was overestimated 3.5 times (predicted: 1.70 % C) using the model derived from calibration set 2. Using the model based on calibration set 1, the prediction of graphite content yielded negative values (predicted: −0.19 % C). In the additionally used artificial soils 2 and 3, which did not contain CaCO3 or graphite, the graphite contents were underestimated with the model based on calibration set 1 (based on soil) and overestimated with the model based on calibration set 2 (based on quartz). Independent of the two models, the relative difference between them is in both cases 0.44, which resembles the actual graphite content. The graphite content of the graphitic schist was estimated to be 2.44 % C according to calibration set 2, but the model calibrated with set 1 showed better predictions. Where the total C content of the graphitic rock was 0.84 % C, the model of calibration set 1 estimated a graphite content of 0.64 % C (Table 2).
Table 2Overview of the predicted graphitic carbon by the different examined methods.
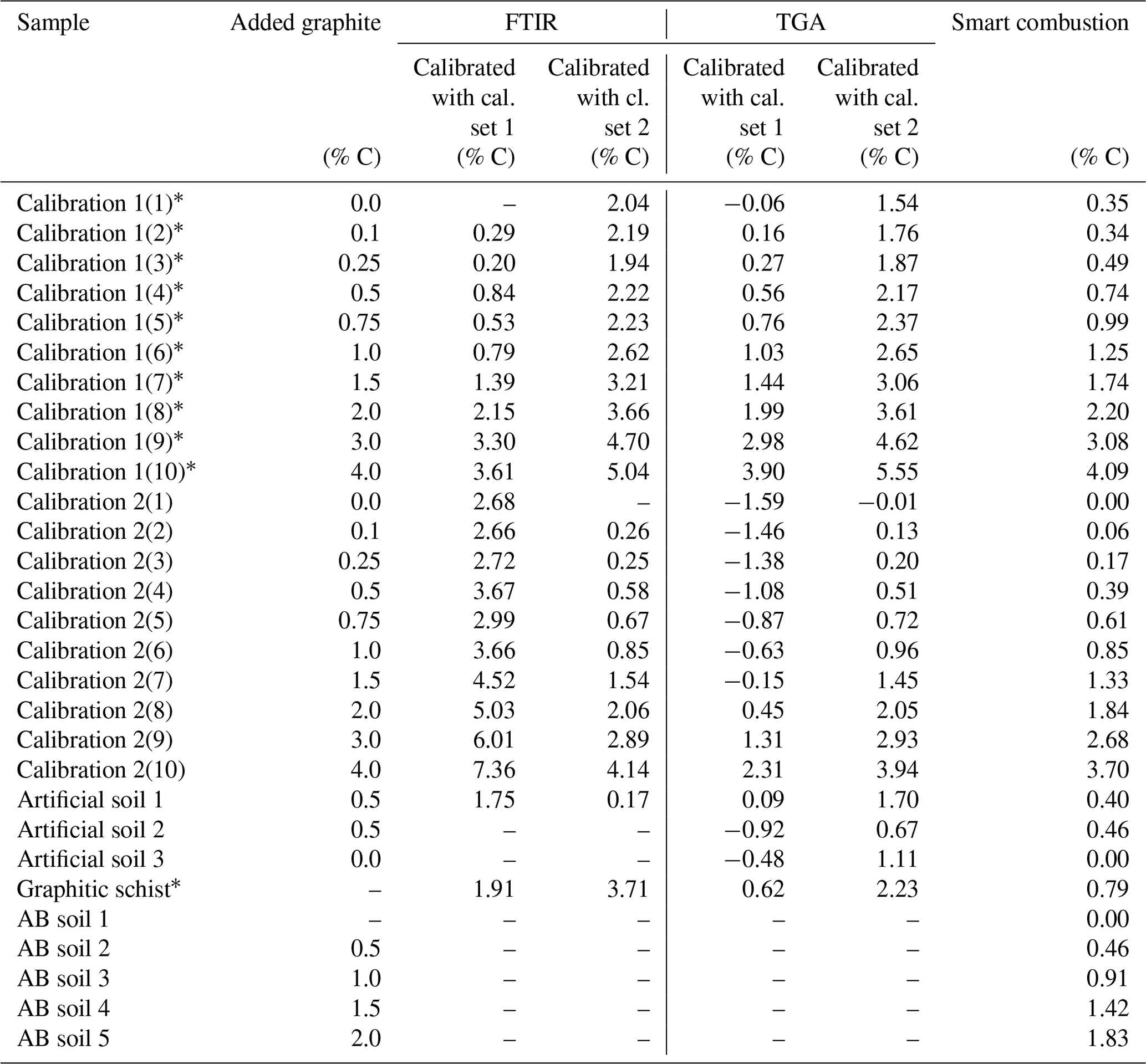
* Contain unknown amount of natural graphite.
Furthermore, the artificial soils were used to explore whether changing between oxic and anoxic conditions during thermal analysis could separate between the mass loss peak of CaCO3 and graphite – most important for potential application in soils containing both components. Changing the atmospheric composition resulted in an artificial mass gain when O2∕N2 gas was switched to N2 and an artificial mass loss when oxygen was reintroduced (Fig. 4), probably due to changes in pressure, and thereby affecting the mass readings. It should be noted that a constant heating program was used and therefore the measurement time spans 4–5 min for both peaks. Nonetheless using artificial soil 2 (without CaCO3) and artificial soil 3 (without graphite) revealed that qualitative separation between the two mass loss peaks was feasible using changes in oxic conditions. Based on TGA observations on some individual components and simplified artificial soil (Fig. 2), it seemed best to use anoxic conditions from 500 till 850 ∘C as all OM will be oxidized at 500 ∘C and carbonates should be fully decomposed at 850 ∘C (Fig. 2). Furthermore, the mass loss peak had already returned to the baseline after reaching 850 ∘C, indicating that the decomposition of the carbonates was complete (Fig. 4).
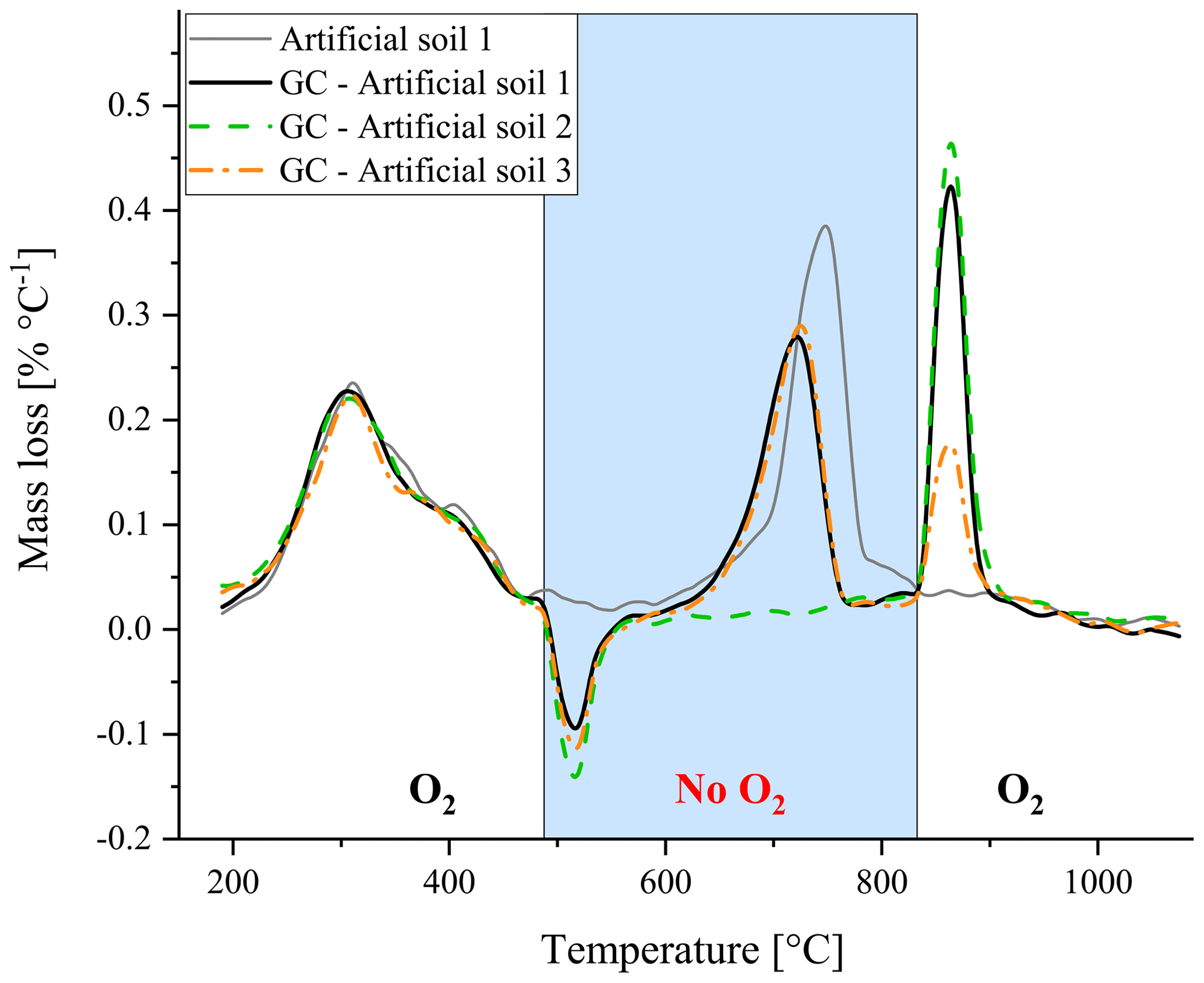
Figure 4Thermogravimetric analysis of the artificial soils, with one lacking either carbonate (artificial soil 2, green) or graphite (artificial soil 3, orange), whereby the oxygen gas supply was cut off during part of the standard heating program (GC – gas change). For comparison the artificial soil 1 under the normal program (without gas change) is also displayed in grey.
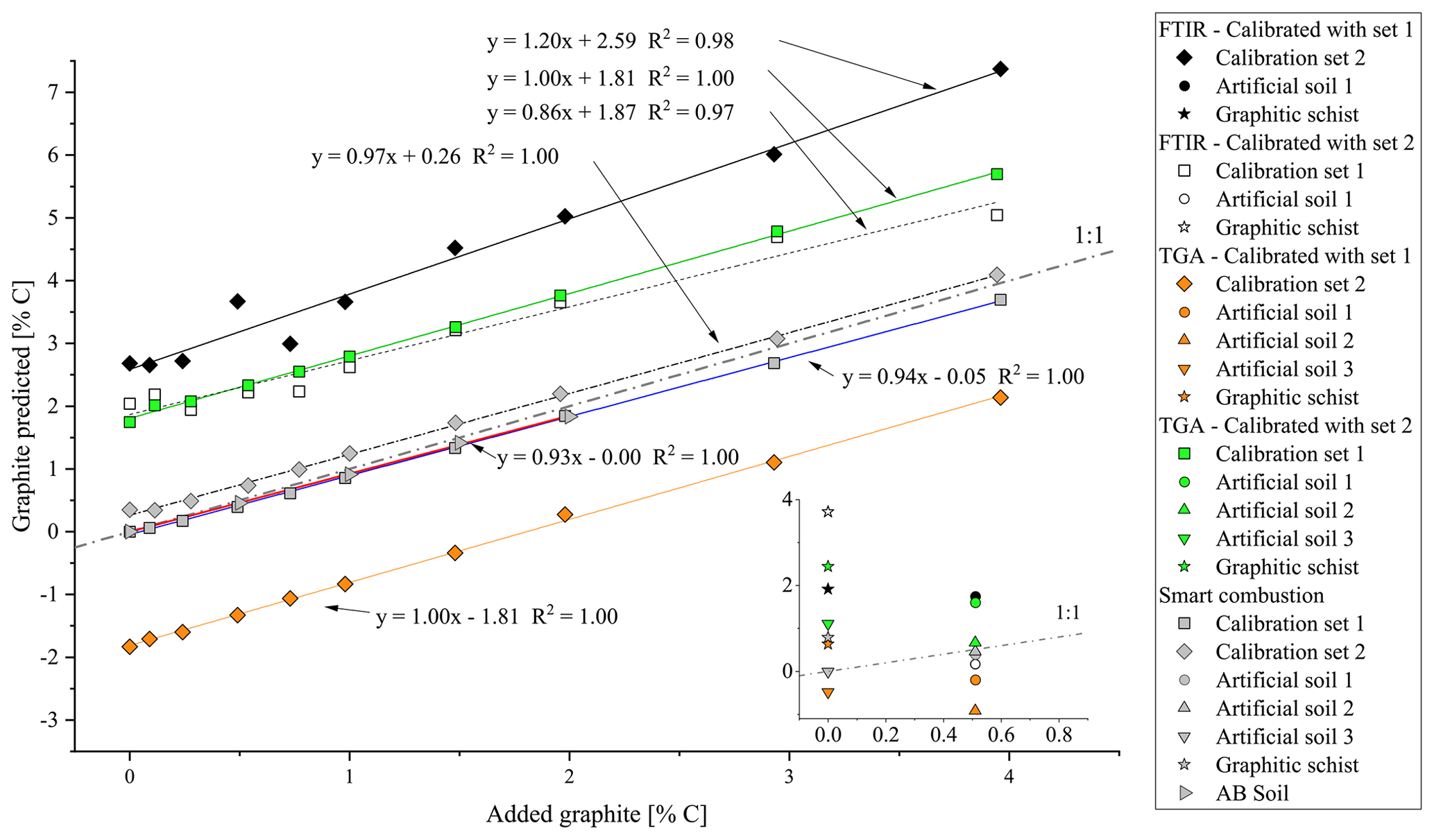
Figure 5Overview of the predicted amount of graphite in the calibration sets (squares/diamonds), artificial soil (inset, circles/triangle), graphitic schist (inset, stars) and AB soil (right-pointing triangle) as measured with the different methods. Black symbols: graphite prediction by FTIR, model from calibration set 1; white: graphite prediction with FTIR, calibration set 2; orange: graphite prediction by TGA, model from calibration set 1; green: graphite prediction by TGA, model from calibration set 2; grey: graphite prediction by smart combustion. Exact data are given in Table 2.
3.3 Direct graphite content quantified by smart combustion
First total carbon (TC) measured by the smart combustion method was compared with the TC obtained by dry combustion using the elemental analyser, but hardly any differences were found (Fig. S5). Residual oxidizable C (ROC) values obtained by the smart combustion method were plotted against the added amount of graphite in calibration sets (Fig. 5). The graphite content in calibration set 1 seems to be overestimated by 0.26 % C. This observation can be explained by the fact that the used sample soil for calibration set 1 contains an unknown amount of natural graphite, which explains a constant overestimation. The content of graphitic C of the soils in calibration set 2 is slightly underestimated, especially with higher graphite concentrations (Fig. 5 and Table 2).
The graphite content of the artificial soil 1 was slightly underestimated (Table 2): 0.40 % ROC for the artificial soil 1 % and 0.46 % ROC for the artificial soil 2, compared to the real contents of 0.50 %. Artificial soil 3 revealed 0.00 % ROC as there was also no graphite in this sample. The graphitic schist had 0.79 % ROC, which was very similar to the total C of the rock (Tables 1 and 2). Furthermore, the AB soil spiked with graphite, as an additional sample without natural graphite but with a high CaCO3 content (1.87 % C), showed a similar underestimation as observed with the calibration samples (Fig. 5).
The ROC results for the natural graphitic soil, also used for calibration set 1 and spiked with CaCO3, revealed a downward trend of measured ROC content with increasing amounts of added CaCO3 (from 0.0 % to 2.5 % added C) down to 0.01 % ROC absolute (Fig. 6). This observation was also evident when taking into account the dilution of the sample by the addition of CaCO3 and explained at least parts of the underestimation of graphite in this sample (Fig. 5).
4.1 Matrix effects and the lack in specific absorption bands hamper graphite quantification via FTIR spectroscopy
The calibration between infrared spectra and graphite contents of the calibration sets yielded promising results (Fig. 1a and b) and could also be used for a cross-validation (Fig. 5). Although the same substrate materials and similar contents of graphitic C were used in the validation, the graphite contents were systematically over-predicted. Despite the apparent quality of the calibration, this failure could have been caused by the relatively low number of calibration samples. Note that the use of the two calibration data sets, soil and quartz, in a joint PLSR model (R2=0.96 and RMSEP = 0.24; three components) did not improve the calibration nor the prediction accuracy. It cannot be excluded that a higher number of samples for the calibration could improve the PLSR model and the prediction results. Further, Raman spectroscopy might be an alternative approach for quantifying graphite in soil samples (e.g. Sparkes et al., 2013; Jorio and Filho, 2016).
The loading values revealed that the relationships used for the PLSR calibration (Fig. 1c and d) could not be attributed to absorption bands of graphite reported in the literature such as wave numbers 2200, 1587, 1362, and 830 cm−1 (Friedel and Carson, 1971) or 3400, 1729, 1220, 1052 and 870 cm−1 (Tan et al., 2013). These wave-number positions did not match with the absorption bands of the spectra obtained from calibration sets 1 and 2, the graphitic schist and the artificial soil 1 (Fig. 7). The only exception is WN 3400 cm−1, which is mainly caused by O–H of free or adsorbed water and is thus not specific for graphite.
The height of the loading values across broad spectral regions, i.e. across the entire wave-number range in the case of the soil samples, hints at effects of the general optical conditions within the samples. The transmission, i.e. the energy throughput in the sample pellet (transmission FTIR) or the reflectance of the sample surface (DRIFT), seems to be a measure for the amount of graphite added rather than specific graphite signal intensities in the calibration sets. In addition, transmission/reflection characteristics are highly influenced by the mineral composition of a sample. Generally, increasing graphite concentrations caused decreasing transmission over the entire spectral range, which is a characteristic of the mineral composition due to increasing proportions of primary absorption (Kortüm, 1969; Hesse et al., 1984). This assumption is underlined by the fact that calibration was not possible with both calibration sets when using baseline-corrected spectra because baseline correction compensated for the described effect of decreasing transmission over the entire wave-number range with increasing graphite content. In addition, DRIFT measurements of pure, i.e. undiluted, graphite material (not shown) did not reveal any prominent absorption bands. Note that specific graphite absorption bands that have been reported in the literature are only valid for oxidized graphite where C–O and C=O groups have been formed to a certain extent (Tan et al., 2013). While FTIR spectroscopy may be feasible for determining oxidized or impure graphite, it was found to be not useful in our case, where an oxidization or impureness of the used graphite material obviously did not appear. Further, the mentioned potential signal intensities would occur in the same spectral ranges as compared to signals from SOM functional groups, thus hampering a quantification of graphite in soil samples. Consequently, the lack of specific absorption bands resulted in a strong dependency of the calibration and validation quality on the sample matrix, i.e. its main mineral component. This matrix effect was illustrated by the incapability of the PLSR models to predict the graphite contents of the artificial soil 1 or graphitic schist (Fig. 5 and Table 2).
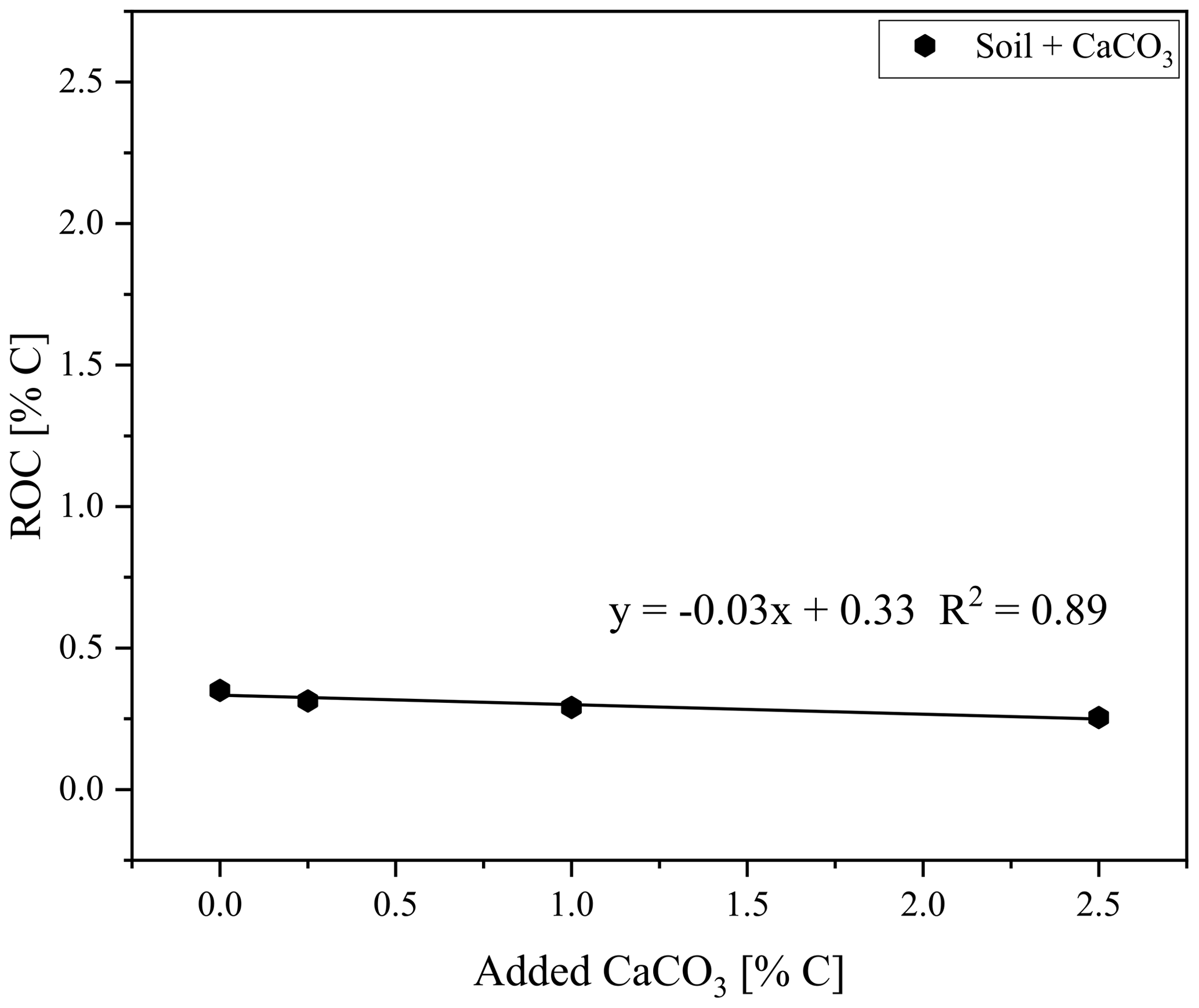
Figure 6Residual oxidizable carbon (ROC) as measured with the smart combustion method plotted against the added CaCO3 content to soil sample, used for creating calibration set 1.
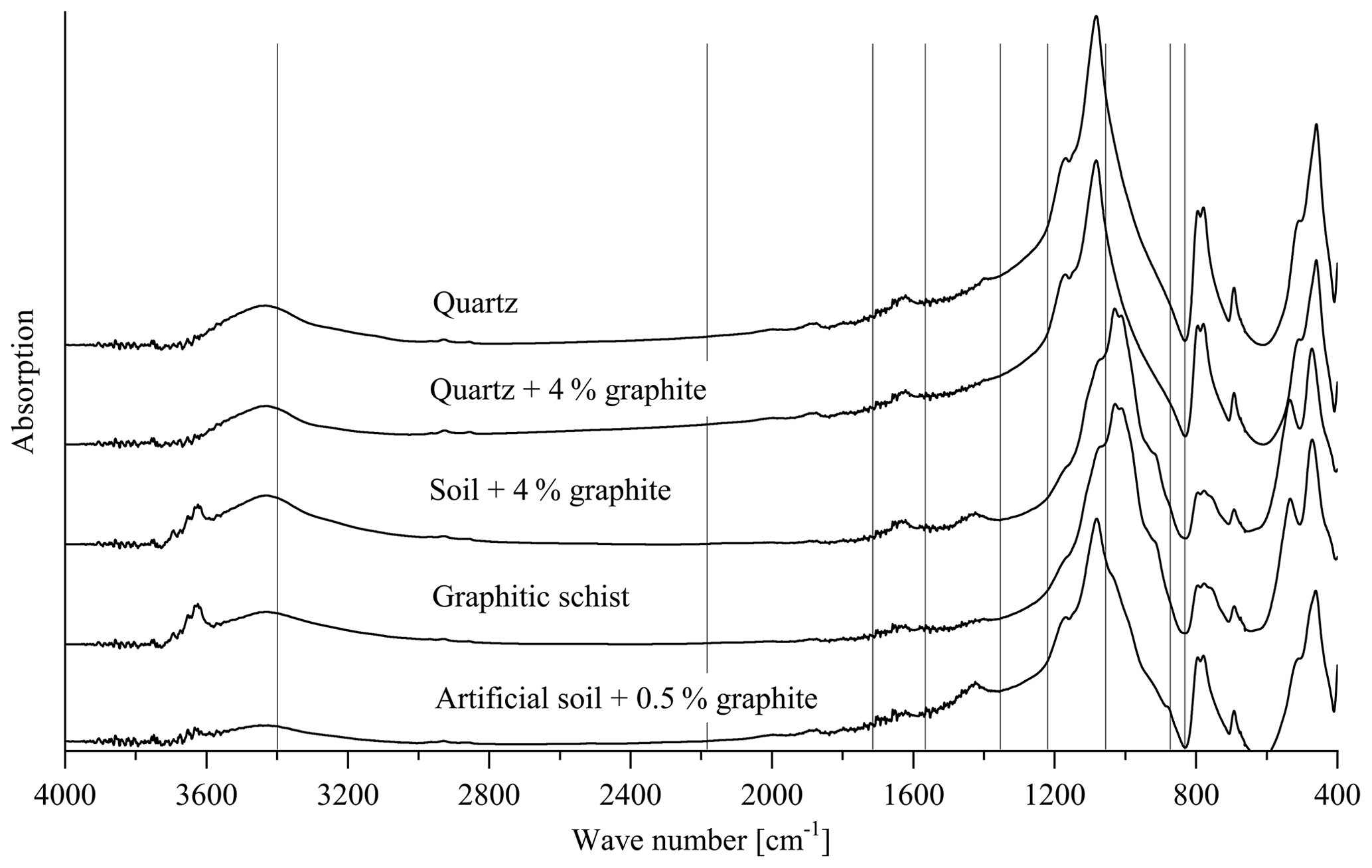
Figure 7Transmission spectra of the pure quartz (calibration set 2, sample 1), quartz +4 % graphite (calibration set 2, sample 10), soil +4 % graphite (calibration set 1, sample 10), artificial soil 1 (0.5 % graphite added) and graphitic schist. The vertical lines denote wave numbers for which absorption peaks have been reported in the literature (see text).
4.2 Strong matrix effects did not allow use of TGA as a universal quantification method
In recent work, the TGA method has been tested and further developed for differentiating between carbonates and OC∕OM (e.g. Apesteguia et al., 2018). First analysis of individual components in a quartz matrix revealed that graphite has a similar thermal stability as carbonates (Fig. 2) and overlap in the thermal region where dehydroxylation of various minerals takes place (Földvári, 2011; Fernaíndez et al., 2012). As shown in Figs. S2 and S3 and discussed by other studies, complete removal of carbonates from soil sample by acid fumigation is difficult and might affect further quantitative estimations. For graphite, estimation via TGA becomes challenging as the acid affects the thermal stability of other soil constituents and makes the sample hygroscopic (Agarwal and Bucheli, 2011; Apesteguia et al., 2018). Additionally, sample grinding in an agate disk mill, representing common homogenization process used for small sample amounts (10s of mg), introduce some changes in thermogravimetric patterns for some minerals, e.g. micas, but makes it also more “reactive” (Földvári, 2011). This would mean that mass loss peaks for minerals, like the used muscovite, can appear sharper and at lower temperatures in comparison with non-ground materials.
The best temperature range to relate mass loss to the amount of added graphite was between 680 and 840 ∘C. For calibration set 2 (pure quartz matrix) a lower temperature (range) would also be able to predict the graphite content (Fig. S4), indicating interferences in the soil matrix of calibration set 1. The best temperature range was in line with the observation that the mass loss peak of graphite spans a large range (Fig. 2), most likely a result of the slow oxidation of this pure C. Other studies found that graphite in a (fluid) sand bed already oxidized slowly under oxygen-rich conditions at temperatures below 670 ∘C accelerating at higher temperatures (Hayhurst et al., 1998; Bews et al., 2001).
Validation of the created models from the two calibration sets revealed that interference with other soil components required an individual calibration for every sample set of specific (mineral) composition (Fig. 5). As shown in this study, fresh but ground muscovite dehydroxylates between 600 and 1000 ∘C (Fig. 2), which influenced the total mass loss measured in this temperature range. Other present (minor) minerals, like chlorites (500–860 ∘C) or apatites (200–1400 ∘C), might also increase the bias by influencing mass loss (Földvári, 2011; Tõnsuaadu et al., 2011). This observation could explain why the model of calibration set 1, using the soil spiked with graphite, showed a good predictability of the graphite content in the graphitic schist, as mineral composition is highly similar between these two samples (Fig. 5 and Table 2).
Roth et al. (2012) suggested that the use of anoxic conditions/a pyrolysing phase during measurement might be useful way to differentiate between wood and black C. According to our gas switching experiment with the TGA, it is at least a useful approach to differentiate between graphite and CaCO3 (Fig. 4). Due to the artificial mass gain/loss induced by switching the gases during the measurement, exact temperature ranges for developing a quantitative method could not be established. As no universally accepted temperature limits for the quantification of TOC, TIC or other carbonaceous substances exist, the best temperature ranges for switching between oxic and anoxic conditions are difficult to define (Pallasser et al., 2013; Ussiri et al., 2014). For instance, according to the DIN19539 standard, TOC is defined as the oxidizable C at a maximum of 400 ∘C. Others showed that 1∕5 or even 1∕3 of the TOC is not oxidized at 400 ∘C (Pallasser et al., 2013; Schiedung et al., 2017). For the artificial soil in our study a temperature limit of 400 ∘C seems to be too low to oxidize all OM, as indicated by the TGA in Fig. 2, and therefore the pyrolysing phase was set to 500 ∘C. To obtain a clear peak for the graphite oxidation, it is important that the other substances, i.e. CaCO3, are already decomposed. In the case of the artificial soil, it was found that at 850 ∘C all CaCO3 was decomposed and a clear peak for graphite was formed upon re-establishing the oxic conditions (Fig. 4). With higher CaCO3 levels or dolomitic carbonates, a higher temperature might be needed to create a clear separation between the substances (Földvári, 2011).
TGA seemed to be a good method to identify different organic components of samples and thus can be used as a complemental technique to other methods for (organic) C content estimation. For high graphite content with negligible amounts of dehydroxylating minerals and/or decomposing carbonates, TGA might be a useful method to quantify graphite.
4.3 Minor effect by CaCO3 and radicals on direct graphite quantification using smart combustion
With the TGA method it was already shown that qualitative differentiation between carbonates and graphite was possible by changing between oxic and anoxic conditions during heating of the sample (Fig. 4). Using the Soli-TOC device, a direct measurement of the released C could be achieved during the heating/gas changing program, which correspond very closely to the amount of (added) graphite (Fig. 5 and Table 2). The fact that the Soli-TOC device measured almost the same TC values as the elemental analyser (Fig. S5) supported the idea that a direct comparison between the ROC fraction and (added) graphite content is possible.
As shown by Hayhurst and Parmar (1998), very small impurities in the graphite can cause a small part of the graphite to pyrolyse during anoxic conditions at higher temperatures. Graphitic C of lesser graphitization might therefore result in a larger loss of graphitic C during pyrolysis and a greater underestimation of the graphitic C content. Taking a closer look at the measurements of the artificial soil reveals that a small part of the graphite began to oxidize under anoxic conditions (Fig. 8). The measurement of graphite in quartz, as done for calibration set 2, showed that about 6 % of the total C was lost during the pyrolysis phase, while for the graphitic schist this loss was 2 % (Fig. 8c), resulting in a small underestimation of the graphitic C content. Bews et al. (2001) suggested that at temperatures higher than 700 ∘C, radicals like HO2 and OH might act as reactants with the pure C. Furthermore, in the method comparison study for recovering different black C types, Roth et al. (2012) suggested a (relatively) strong catalytic effect of oxides on black C oxidation, which was most predominant in soils. These ideas are also supported by our observation that artificial soil 2 (without CaCO3) measured higher ROC values (0.06 % more C absolute) than artificial soil 1 (with CaCO3, Table 2). Also in the carbonate-rich AB soil the added graphite was underestimated by 7 % (Fig. 5). Furthermore, graphitic C was underestimated with increasing CaCO3 content (Fig. 6). The 7 % underestimation by the AB soil, which contains 1.87 % C–CaCO3, coincided with ROC underestimation of the calibration soil with 2 %C–CaCO3 addition.

Figure 8Examples of smart combustion measurements of the artificial soils (a), the fresh OM component (b), and the graphite standard and graphitic schist (c). The blue area delineates the interval where O2 gas is substituted for N2. The temperature program is displayed by the red dashed line. Note that artificial soil 2 (green) is without CaCO3 and artificial soil 3 (orange) is without graphite.
When the soil contains more thermally resistant OM, which is not oxidizable at 400 ∘C and can be 1∕4 of the OM (Schiedung et al., 2017), the question arises whether this fraction is pyrolysed during heating under anoxic conditions or whether it is taken as part of the ROC fraction when oxygen is again available. The TGA method showed that not all OM has been oxidized at 400 ∘C (Fig. 2). Taking a closer look at the smart combustion measurement of the artificial soil 2 and particularly its fresh OM component (Fig. 8) revealed that a small peak formed upon heating the sample above 400 ∘C. This peak represents only a few percent of the total OC, but clearly indicates that OM was oxidized incompletely at 400 ∘C. Although this study focuses on the ROC component, this point might be important when considering the TOC400 and TIC fractions of the smart combustion method.
When a sample contains other forms of thermally resistant OM or even black C, which are not pyrolysed during the anoxic phase, this C component is likely to end up in the graphitic C fraction with the smart combustion method. Especially as most temperature boundaries are empirically derived (Pallasser et al., 2013; Ussiri et al., 2014), a pre-test with continuous heating under oxic conditions is therefore recommended to assess the number of C-containing substances present in the sample by the occurrence of peaks. Further studies should focus on temperature boundaries of different substances in relation to their properties and see, for instance, how graphitic C can be distinguished from other thermally stable C components.
Although the thermal boundaries for the different C fractions given in the DIN19539 (GS) standard are debatable (Ussiri et al., 2014; Schiedung et al., 2017), we showed that the ROC fraction corresponded closely to the graphitic C content. Through the smart combustion method, graphitic C could be differentiated from the other C components in the soil matrix and quantified satisfactorily, as indicated by the offset in the calibration with graphite estimation in the artificial/spiked samples.
4.4 Potential for combining methods
Comparing the ability of the examined methods on predicting graphite content, it becomes clear that FTIR overestimated, TGA was highly variable, and smart combustion was most accurate in predicting the graphite content (Fig. 5). An interesting observation was the similar predicted graphite content in calibration set 1 by both the FTIR and TGA methods, especially as FTIR is based on spectral properties and TGA on the thermal stability of the graphite. It has previously been suggested to combine FTIR and TGA systems to rapidly characterize the soil OM (Demyan et al., 2013). Oxidation of graphite upon heating could result in specific infrared absorption bands (Tan et al., 2013), which nevertheless would still be superimposed by SOM-specific bands in natural soil samples. As discussed by Demyan et al. (2013), not only the available oxygen but also the heating rate has an important effect on the charring of OM and thereby on the thermal and spectral properties of the studied material.
As Raman spectroscopy is suitable for distinguishing graphitic C and determining its degree of graphitization, it seems to be a promising method. Nonetheless, to use Raman spectroscopy for quantification of substances in a soil matrix, further studies should first focus on standardization of sample preparation, as it has a large influence on the measured intensities and baseline determination and thereby the direct quantification of components (Beysacc and Lazzeri, 2012; Sparkes et al., 2013).
Because we focused in this study on the ROC component, which significantly correlated with the graphite content, a consideration of other components mentioned in the DIN19539 standard was beyond the scope of this study. Nonetheless, we found indications that the thermal boundaries defined in the DIN19539 standard are not ideal for differentiating between soil OM and inorganic C (Fig. 8). As most carbonates start to decompose at temperatures of 550 ∘C (Földvári, 2011), it might be more suitable to increase the temperature level from 400 to 500 ∘C for the TOC component. Only when black C, which might oxidize between 375 and 540 ∘C (Roth et al., 2012), is present in the sample might this lead to an overestimation of the TOC content. Using TGA simultaneously with differential scanning calorimetry, CO2∕H2O flux measurements (i.e. evolved gas analysis, Fernández et al., 2012) or the Rock-Eval method focusing on hydrocarbon, CO2 and CO release (Behar et al., 2001) could improve the development of a more standardized method applicable to soils using combustion elemental analysers. The overlap between the thermal properties of different C components emphasizes the need to consider the potential as well as possible interferences with the considered methodology before applying a fast and standardized analytic method.
Three widely used methods were examined for their potential to quantify graphitic C content in soil samples. Calibrations between mid-infrared transmission as well as DRIFT spectra and graphite contents of well-defined samples are principally possible via PLSR. However, these calibrations depend on general effects of graphite contents on the energy transmitted through the samples rather than on signal intensities of specific graphite absorption bands. The use of samples from different origins yield strong matrix effects and hampers the prediction of geogenic graphite contents in soils. Thermogravimetric analysis of the samples revealed that the suitability of this qualitative method for identifying graphitic C in soil samples, although care should be taken for carbonates as they have similar thermal stability. Quantitative estimation of the graphite content seems challenging as dihydroxylation of several soil minerals occur at similar temperatures, requiring the calibration with an empirical model. With alteration between oxic and anoxic conditions during heating of a sample, a differentiation between other soil components and graphite could be established, like applied in the smart combustion method. Further quantification of the released C during the gas changing heating program revealed a close correspondence between the measured ROC and original graphite content. Among the examined methods, the smart combustion method performs best in differentiating between graphite and other soil components and thereby also in quantifying graphitic C in soil samples.
Data underlying the figures and tables are accessible via https://doi.org/10.25532/OPARA-48. Additionally, FTIR spectra and TGA Thermograms are available via respectively https://doi.org/10.25532/OPARA-49 and https://doi.org/10.25532/OPARA-47 (Zethof, 2019).
The supplement related to this article is available online at: https://doi.org/10.5194/soil-5-383-2019-supplement.
ML contributed the FTIR part to the article, including analysis of the data and editing of the paper. CV contributed by suggesting and evaluating a test set-up and by commenting on and editing the paper. SS contributed by providing measurements with the Soli-TOC device, technical assistance and editing of the paper. KK contributed by suggesting and evaluating the test set-up and by commenting on and editing the paper.
The authors declare that they have no conflict of interest.
We thank Manuela Unger and Gisela Ciesielski for the laboratory assistance and the (anonymous) reviewers for their helpful input. We also want to thank Johanna Hartmann and the Leibniz Institute (Dresden) for providing the Raman spectra.
This research has been supported by the Deutsche Forschungsgemeinschaft (DFG) (grant nos. KA1737/13-1, LE3177/1-2), and Eropean Union's Horizon 2020 programme (grant no. 695101).
This paper was edited by Carolina Boix-Fayos and reviewed by two anonymous referees.
Agarwal, T. and Bucheli, T. D.: Adaptation, validation and application of the chemo-thermal oxidation method to quantify black carbon in soils, Environ. Pollut., 159, 532–538, https://doi.org/10.1016/j.envpol.2010.10.012, 2011.
Apesteguia, M., Plante, A. F., and Virto, I.: Methods assessment for organic and inorganic carbon quantification in calcareous soils of the Mediterranean region, Geoderma Regional, 12, 39–48, https://doi.org/10.1016/j.geodrs.2017.12.001, 2018.
Barré, P., Plante, A. F., Cécillon, L., Lutfalla, S., Baudin, F., Bernard, S., Christensen, B. T., Eglin, T., Fernandez, J. M., Houot, S., Kätterer, T., Le Guillou, C., Macdonald, A., van Oort, F., and Chenu, C.: The energetic and chemical signatures of persistent soil organic matter, Biogeochemistry, 130, 1–12, https://doi.org/10.1007/s10533-016-0246-0, 2016.
Behar, F., Beaumont, V., and Penteado, H. D. B.: Rock-Eval 6 technology: performances and developments, Oil Gas Sci. Technol., 56, 111–134, https://doi.org/10.2516/ogst:2001013, 2001.
Bews, I. M., Hayhurst, A. N., Richardson, S. M., and Taylor, S. G.: The order, Arrhenius parameters, and mechanism of the reaction between gaseous oxygen and solid carbon, Combust. Flame, 124, 231–245, https://doi.org/10.1016/S0010-2180(00)00199-1, 2001.
Beyssac, O. and Rumble, D.: Graphitic carbon: a ubiquitous, diverse, and useful geomaterial, Elements, 10, 415–420, https://doi.org/10.2113/gselements.10.6.415, 2014.
Blankinship, J. C., Berhe, A. A., Crow, S. E., Druhan, J. L., Heckman, K. A., Keiluweit, M., Lawrence, C. R., Marín-Spiotta, E., Plante A. F., Rasmussen, C., Schädel, C., Schimel, J. P., Sierra, C. A., Thompson, A., Wagai, R., Wieder, W. R., and Schädel, C.: Improving understanding of soil organic matter dynamics by triangulating theories, measurements, and models, Biogeochemistry, 140, 1–13, https://doi.org/10.1007/s10533-018-0478-2, 2018.
Buseck, P. R. and Beyssac, O.: From organic matter to graphite: Graphitization, Elements, 10, 421–426, https://doi.org/10.2113/gselements.10.6.421, 2014.
Clark, K. E., Hilton, R. G., West, A. J., Robles Caceres, A., Gröcke, D. R., Marthews, T. R., Ferguson, R. I., Asner, G. P., New, M., and Malhi, Y.: Erosion of organic carbon from the Andes and its effects on ecosystem carbon dioxide balance, J. Geophys. Res.-Biogeo., 122, 449–469, https://doi.org/10.1002/2016JG003615, 2017.
Chung, D. D. L.: Review Graphite, J. Mat. Sci., 37, 1475–1489, 2002.
Demyan, M. S., Rasche, F., Schütt, M., Smirnova, N., Schulz, E., and Cadisch, G.: Combining a coupled FTIR-EGA system and in situ DRIFTS for studying soil organic matter in arable soils, Biogeosciences, 10, 2897–2913, https://doi.org/10.5194/bg-10-2897-2013, 2013.
DIN Standards Committee Water Practice, Investigation of solids – Temperature-dependent differentiation of total carbon (TOC400, ROC, TIC900), Beuth, Berlin, Germany, available at: https://www.din.de/en/getting-involved/standards-committees/naw/standards/wdc-beuth:din21:263898867 (last access: 8 April 2019), 2016.
Fernaìndez, J. M., Peltre, C., Craine, J. M., and Plante, A. F.: Improved characterization of soil organic matter by thermal analysis using CO2∕H2O evolved gas analysis, Environ. Sci. Technol., 46, 8921–8927, https://doi.org/10.1021/es301375d, 2012.
Ferrari, A. C.: Raman spectroscopy of graphene and graphite: Disorder, electron–phononcoupling, doping and nonadiabatic effects, Solid State Commun., 143, 47–57, https://doi.org/10.1016/j.ssc.2007.03.052, 2007.
Földvári, M.: Handbook of the thermogravimetric system of minerals and its use in geological practice, Budapest, Hungary, Geo. Inst. Hungary, 213, 180 pp., 2011.
Friedel, R. A. and Carlson, G. L.: Infrared Spectra of Ground Graphite, J. Phys. Chem., 75, 1149–1151, 1971.
Galvez, M. E., Beyssac, O., Martinez, I., Benzerara, K., Chaduteau, C., Malvoisin, B., and Malavieille, J.: Graphite formation by carbonate reduction during subduction, Nat. Geosci., 6, 473–477, https://doi.org/10.1038/NGEO1827, 2013.
Galy, V., Beyssac, O., France-Lanord, C., and Eglinton, T.: Recycling of graphite during Himalayan erosion: a geological stabilization of carbon in the crust, Science, 322, 943–945, https://doi.org/10.1126/science.1161408, 2008.
Harris, D., Horwáth, W. R., and Van Kessel, C.: Acid fumigation of soils to remove carbonates prior to total organic carbon or carbon-13 isotopic analysis, Soil Sci. Soc. Am. J., 65, 1853–1856, https://doi.org/10.2136/sssaj2001.1853, 2001.
Hartmann, J. and Moosdorf, N.: The new global lithological map database GLiM: A representation of rock properties at the Earth surface. Geochem. Geophy. Geosy., 13, Q12004, https://doi.org/10.1029/2012GC004370, 2012.
Hayhurst, A. N. and Parmar, M. S.: Does solid carbon burn in oxygen to give the gaseous intermediate CO or produce CO2 directly? Some experiments in a hot bed of sand fluidized by air, Chem. Eng. Sci., 53, 427–438, https://doi.org/10.1016/S0009-2509(97)00334-5, 1998.
Hemingway, J. D., Hilton, R. G., Hovius, N., Eglinton, T. I., Haghipour, N., Wacker, L., Chen, M-C., and Galy, V. V.: Microbial oxidation of lithospheric organic carbon in rapidly eroding tropical mountain soils, Science, 360, 209–212, https://doi.org/10.1126/science.aao6463, 2018.
Hesse, M., Meier, H., and Zeeh, B.: Spectroscopic methods in organic chemistry, Georg Thieme, Stuttgart, Germany, 56–74, 1984 (in German).
Instituto Geologico y Minero de España (IGME): Mapa Geologico de España I, 50 000 – Hoja Tabernas (Madrid: Ministerio de Industria y Energia), available at: http://info.igme.es/cartografiadigital/geologica/Magna50Hoja.aspx?language=en{&}id=1030 (last access: 14 March 2019), 1975.
Instituto Geologico y Minero de España (IGME): Mapa Geologico de España Escala 1:50.000 – Hoja Gergal (Madrid: Ministerio de Industria y Energia), available at: http://info.igme.es/cartografiadigital/geologica/Magna50Hoja.aspx?language=en{&}id=1029 (last access: 14 March 2019), 1979.
Janik, L. J., Merry, R. H., and Skjemstad, J. O.: Can mid infrared diffuse reflectance analysis replace soil extractions?, Aust. J. Exp. Agr., 38, 681–694, 1998.
Jorio, A. and Filho, A. G. S.: Raman Studies of Carbon Nanostructures, Annu. Rev. Mater. Res., 46, 357–382, https://doi.org/10.1146/annurev-matsci-070115-032140, 2016.
Kortüm, G.: Reflexionsspektroskopie, Springer-Verlag, Berlin, 378 pp., 1969.
Kučerík, J., Tokarski, D., Demyan, M. S., Merbach, I., and Siewert, C.: Linking soil organic matter thermal stability with contents of clay, bound water, organic carbon and nitrogen, Geoderma, 316, 38–46, https://doi.org/10.1016/j.geoderma.2017.12.001, 2018.
Liu, L., Zhu, C., Fan, M., Chen, C., Huang, Y., Hao, Q., Yang, J., Wang, H., and Sun, D.: Oxidation and degradation of graphitic materials by naphthalene-degrading bacteria, Nanoscale, 7, 13619–13628, https://doi.org/10.1039/c5nr02502h, 2015.
McCarty, G. W., Reeves, J. B., Reeves, V. B., Follett, R. F., and Kimble, J. M.: Mid-Infrared and Near-Infrared Diffuse Reflectance Spectroscopy for Soil Carbon Measurement, Soil Sci. Soc. Am. J., 66, 640–646, 2002.
Mevik, B. H., Wehrens, R., and Liland, K. H.: pls: Partial Least Squares and Principal Component Regression, R package version 2.7-0, available at: https://CRAN.R-project.org/package=pls (last accss: 1 April 2019), 2018.
Oohashi, K., Hirose, T., Kobayashi, K., and Shimamoto, T.: The occurrence of graphite-bearing fault rocks in the Atotsugawa fault system, Japan: Origins and implications for fault creep, J. Struct. Geol., 38, 39–50, https://doi.org/10.1016/j.jsg.2011.10.011, 2012.
Pallasser, R., Minasny, B., and McBratney, A. B.: Soil carbon determination by thermogravimetrics, Peer J., 1, e6, https://doi.org/10.7717/peerj.6, 2013.
Parikh, S. J., Goyne, K. W., Margenot, A. J., Mukome, N. D., and Calderon, F. J.: Soil chemical insights provided through vibrational spectroscopy, Adv. Agron., 126, 1–148, https://doi.org/10.1016/B978-0-12-800132-5.00001-8, 2014.
Plante, A. F., Fernández, J. M., and Leifeld, J.: Application of thermal analysis techniques in soil science, Geoderma, 153, 1–10, https://doi.org/10.1016/j.geoderma.2009.08.016, 2009.
Puigdefábregas, J., Alonso, J. M., Delgado, L., Domingo, F., Cueto,M., Gutiérrez, L., Lázaro, R., Nicolau, J. M., Sánchez, G., Solé, A., Vidal, S., Aguilera, C., Bremner, A., Clarks, S., and Incoll, L.: The Rambla Honda field site: interactions of soil and vegetation along a catena in semiarid southeast Spain, in: Mediterranean Desertification and Land use, edited by: Brandt, J. and Thornes, J. B., Wiley & Sons, Chinchester, England, 137–167, 1996.
R Development Core Team: R: A Language and Environment for Statistical Computing, R Foundation for Statistical Computing, Vienna, Austria, available at: https://www.r-project.org/ (last access: 7 December 2019), 2014.
R Development Core Team: R: A Language and Environment for Statistical Computing, R Foundation for Statistical Computing, Vienna, Austria, available at: https://www.r-project.org/ (last access: 7 December 2019), 2018.
Reeves, J. B.: Mid-infrared diff use reflectance spectroscopy: Is sample dilution with KBr necessary, and if so, when?, Am. Lab., 35, 24–25, 2003.
Reeves, J. B., McCarty, G., and Mimmo, T.: The potential of diffuse reflectance spectroscopy for the determination of carbon inventories in soils, Environ. Pollut., 116, 277–284, 2002.
Roth, P. J., Lehndorff, E., Brodowski, S., Bornemann, L., Sanchez-García, L., Gustafsson, Ö., and Amelung, W.: Differentiation of charcoal, soot and diagenetic carbon in soil: Method comparison and perspectives, Org. Geochem., 46, 66–75, https://doi.org/10.1016/j.orggeochem.2012.01.012, 2012.
Rumble, D.: Hydrothermal graphitic carbon, Elements, 10, 427–433, https://doi.org/10.2113/gselements.10.6.427, 2014.
Rumpel, C. and Kögel-Knabner, I.: Deep soil organic matter – a key but poorly understood component of terrestrial C cycle, Plant Soil, 338, 143–158, https://doi.org/10.1007/s11104-010-0391-5, 2011.
Sadezky, A., Muckenhuber, H., Grothe, H., Niessner, R., and Pöschl, U.: Raman microspectroscopy of soot and related carbonaceous materials: spectral analysis and structural information, Carbon, 43, 1731–1742, https://doi.org/10.1016/j.carbon.2005.02.018, 2005.
Schiedung, M., Don, A., Wordell-Dietrich, P., Alcántara, V., Kuner, P., and Guggenberger, G.: Thermal oxidation does not fractionate soil organic carbon with differing biological stabilities, J. Plant Nutr. Soil Sc., 180, 18–26, https://doi.org/10.1002/jpln.201600172, 2017.
Schmidt, M. W., Skjemstad, J. O., and Jäger, C.: Carbon isotope geochemistry and nanomorphology of soil black carbon: Black chernozemic soils in central Europe originate from ancient biomass burning, Global Biogeochem. Cy., 16, 1123, https://doi.org/10.1029/2002GB001939, 2002.
Senesi, N., D'Orazio, V., and Ricca, G.: Humic acids in the first generation of EUROSOILS, Geoderma, 116, 325–344, https://doi.org/10.1016/S0016-7061(03)00107-1, 2003.
Smith, B. C.: Fundamentals of Fourier Transform Infrared Spectroscopy, CRC press, p. 224, 1995.
Sparkes, R., Hovius, N., Galy, A., Kumar, R. V., and Liu, J. T.: Automated analysis of carbon in powdered geological and environmental samples by Raman Spectroscopy, Appl. Spectrosc., 67, 779–788, https://doi.org/10.1366/12-06826, 2013.
Tan, L.-L., Ong, W.-J., Chai, S.-P., and Mohamed, A. R.: Reduced graphene oxide-TiO2 nanocomposite as a promising visible-light-active photocatalyst for the conversion of carbon dioxide, Nanoscale Lett., 8, p. 465, 2013.
Tõnsuaadu, K., Gross, K. A., Plūduma, L., and Veiderma, M.: A review on the thermal stability of calcium apatites, J. Therm. Anal. Calorim., 110, 647–659, https://doi.org/10.1007/s10973-011-1877-y, 2011.
Trumbore, S.: Age of soil organic matter and soil respiration: radiocarbon constraints on belowground C dynamics, Ecol. Appl., 10, 399–411, https://doi.org/10.1890/1051-0761(2000)010[0399:AOSOMA]2.0.CO;2, 2000.
Ussiri, D. A., Jacinthe, P. A., and Lal, R.: Methods for determination of coal carbon in reclaimed minesoils: a review, Geoderma, 214, 155–167, https://doi.org/10.1016/j.geoderma.2013.09.015, 2014.
Van der Marel, H. W. and Beutelspacher, H.: Atlas of Infrared Spectroscopy of Clay Minerals and their Admixtures, Elsevier, Amsterdam, the Netherlands, p. 65, 1976.
Vohland, M., Ludwig, M., Thiele-Bruhn, S., and Ludwig, B.: Determination of soil properties with visible to near- and mid-infrared spectroscopy: Effects of spectral variable selection, Geoderma, 223, 88–96, https://doi.org/10.1016/j.geoderma.2014.01.013, 2014.
WRB (World Reference Base for Soil Resources 2014): IUSS Working Group WRB, International soil classification system for naming soils and creating legends for soil maps, World Soil Resources Reports No. 106. FAO, Rome, 181 pp., 2014.
Zamanian, K., Pustovoytov, K., and Kuzyakov, Y.: Pedogenic carbonates: Forms and formation processes, Earth-Sci. Rev., 157, 1–17, https://doi.org/10.1016/j.earscirev.2016.03.003, 2016.
Zethof, J. H. T.: Accompanying materials for the publication: “Identifying and quantifying geogenic organic carbon in soils – the case of graphite” – Data underlying figures and tables, TGA thermograms, FTIR spectra, https://doi.org/10.25532/OPARA-48, https://doi.org/10.25532/OPARA-47, https://doi.org/10.25532/OPARA-49, 2019.